
Albinismo. Fenotipo e Genotipo
Initial posting: 4 ottobre 2007
last update: aggiornamenti in corso
Sommario
Definizione di Albinismo
Fenotipo albinotico
Fenotipo pigmentario albinotico
Fenotipo oftalmologico albinotico
Genotipo albinotico
Bibliografia
Nota
Sommario
E’ importante dare una definizione di Albinismo, al fine di comprendere la differenza tra il vero Albinismo e gli altri disordini che presentano nel loro fenotipo una condizione clinica di ipopigmentazione, o generalizzata (Sindrome di Griscelli) o localizzata (Piebaldismo e Sindromi di Waardenburg), senza coinvolgimento oculare.Il termine Albinismo è usato per definire anomalie genetiche, caratterizzate da una riduzione congenita della sintesi della melanina, associata a determinati cambiamenti oculari, che sono il risultato di una ridotta quantità di melanina durante lo sviluppo dell’occhio. I melanociti sono normali in numero e struttura, variano invece il grado di melanizzazione o le dimensioni dei melanosomi. Il termine Albinismo Oculocutaneo (OCA) distingue le condizioni caratterizzate da ridotta sintesi di melanina nella cute, nei capelli, nei peli e negli occhi, con grado variabile di melanizzazione, mentre il termine Albinismo Oculare (OA) distingue quella caratterizzata da una ridotta sintesi di melanina fondamentalmente nell’epitelio pigmentato della retina, con presenza di macromelanosomi in tutti i tessuti interessati. I cambiamenti oculari, di entità variabile e non tutti sempre presenti, si ritrovano in entrambe le forme di Albinismo.Il Fenotipo Albinotico presenta un’ eterogeneità clinica molto estesa, con manifestazioni cutanee ed oculari che non sono, in generale, né essenziali, né specifiche dell’Albinismo. Sembra possa considerarsi caratteristica specifica dell’Albinismo, indipendentemente dal genotipo e dal fenotipo, il solo misrouting ottico.Fenotipo pigmentario albinotico. La cute si presenta normale da un punto di vista anatomico-funzionale; cambia solo il colore, che copre uno spettro pigmentario estremamente ampio, in relazione al quale varia la risposta della cute al sole, ossia l’entità dello schermo di protezione naturale. I capelli hanno una normale struttura anatomica, varia solo il colore: dal bianco alle diverse tonalità di biondo e di marrone. Il colore dell’iride (stoma irideo) varia dal blu/grigio opaco al verde/verde nocciola al marrone/marrone chiaro.Fenotipo oftalmologico albinotico. Quando ad un’ ipopigmentazione iridea stromale si aggiunge un’ipopigmentazione dell’epitelio pigmentato dell’iride (IPE), il risultato è una trasparenza di grado variabile: un’ iride blu, ma opaca, non è albinotica; un’ iride blu e trasparente può essere albinotica; un iride con aree di trasparenza non è necessariamente albinotica. L’iride delle femmine portatrici di OA1, presenta, nell’80-90% dei casi, aree di trasparenza. Un iride trasparente permette alla luce dispersa di entrare e alla luce riflessa dalla retina di uscire, causando ipersensibilità patologica alla luce: fotofobia. La funzione diaframmatica dell’iride non è compromessa. La retina albinotica si presenta rosa/rossastra: la sua trasparenza, determinata dalla riduzione/assenza di melanina nell’epitelio pigmentato della retina, rende visibili i vasi sanguigni corioidei sottostanti. La retina delle femmine portatrici di OA1 X-linked mostra, nell’80-90% dei casi, i “segni del portatore”: depigmentazione irregolare del fondo oculare. La fovea albinotica è ipoplastica, causando la riduzione dell’acuità visiva, non correggibile con l’uso di lenti. Non viene intaccata la visione dei colori, che, nell’albinismo, risulta normale.. Le fibre nervose della retina temporale, giunte a livello del chiasma ottico, decussano, in gran parte, e si proiettano al nucleo genicolato controlaterale, determinando una disorganizzazione e frammentazione del modello laminare dei nuclei genicolati dorso-laterali (LGNs) ed una disorganizzazione delle proiezioni dirette da questi nuclei alla corteccia visiva. Non si ha, di conseguenza sovrapposizione dei campi visivi, con riduzione della funzione binoculare, necessaria per la visone spaziale (stereoscopica), che risulta quindi ridotta o assente. Il nistagmo congenito è un movimento orizzontale involontario degli occhi che compromette la qualità della visione. Di tipo pendolare o a scatti, è a volte associato ad un “ciondolare del capo” o ad una “inclinazione del capo”, che sono in grado di ridurre l’entità del movimento oculare, migliorando, di conseguenza, l’acuità visiva. Presente in quasi tutti gli albini, si manifesta, di solito, nei primi tre mesi di vita. Lo strabismo alternante consiste nella soppressione, intermittente ed alternante, della visione in ogni occhio. Con deviazioni sia verticali che orizzontali, non sviluppa ambliopia e non richiede generalmente (se non per motivi estetici), correzione chirurgica. Frequente è la presenza di miopia o ipermetropia e astigmatismo, errori di rifrazione, determinati dalla forma anomala dell’occhio, assunta nel tentativo di migliorare la qualità dell’immagine sulla retina.Genotipo albinotico. L’eterogeneità fenotipica è associata ad eterogeneità genica e ad eterogeneità allelica. I geni della pigmentazione, finora associati all’Albinismo, sono 14..L’individuazione di soggetti clinicamente albini in cui non è stata rilevata alcuna mutazione in nessuno dei geni noti essere associati all’albinismo, porta a supporre la presenza di mutazioni in regioni regolative o introniche, non ancora esplorate, degli stessi, o l’esistenza di altri geni, non ancora noti, associati a tale fenotipo.
Definizione di Albinismo
E’ importante dare una definizione di Albinismo, al fine di comprendere la differenza tra il vero Albinismo e gli altri disordini che presentano nel loro fenotipo una condizione clinica di ipopigmentazione, o generalizzata o localizzata, senza coinvolgimento oculare.
La Sindrome di Griscelli, autosomica recessiva, è un esempio di ipopigmentazione cutanea generalizzata, determinata da mutazioni a carico del gene MYO5a che codifica la proteina miosina Va, coinvolta, come abbiamo già visto, nel trasporto degli organelli .I melanociti risultano pieni di melanosomi (pigmentati e di misura normale), in quanto pochi di essi vengono trasferiti ai cheratinociti.
Esempi di ipopigmentazione cutanea localizzata sono il Piebaldismo e le Sindromi di Waardenburg. Il Piebaldismo, autosomico dominante, è dovuto a mutazioni del gene KIT, il cui prodotto, recettore di un fattore di crescita, è responsabile dello sviluppo e/o della migrazione dei melanociti. L’aploinsufficienza della trasduzione del segnale determina la mancanza di melanociti in ciocche di peli/capelli bianchi e nelle aree cutanee sottostanti. E’ probabile che anche il suo ligando, se difettoso, determini lo stesso fenotipo. Mutazioni a carico del gene corrispondente, MCF (fattore di crescita delle mastcellule), non sono state ancora riportate. Le Sindromi di Waardenburg di tipo I e III, autosomiche dominanti, sono causate da mutazioni a livello del gene PAX3, il cui prodotto, un fattore di trascrizione transattivante, regola, in combinazione con il prodotto del gene SOX10, l’espressione di diversi geni bersaglio, tra cui il gene MITF. Quest’ultimo codifica un fattore di trascrizione (con una struttura b-HLH-ZIP : dominio basico-dominio helix-loop-helix-dominio leucine-zipper), che, regola, sotto il controllo coordinato della via della PKA cinasi e della via della MAP cinasi, l’espressione dei geni melanogenici (vedi regolazione della melanogenesi). Mutato, determina la Sindrome di Waardenburg di tipo II (autosomica dominante), per aploinsufficienza del prodotto proteico, importante nello sviluppo e nella sopravvivenza dei melanociti.
Il termine Albinismo è usato per definire anomalie genetiche, caratterizzate da una riduzione congenita della sintesi della melanina, associata a specifici cambiamenti oculari, che sono il risultato di una ridotta quantità di melanina durante lo sviluppo dell’occhio. I melanociti sono normali in numero e struttura, variano invece il grado di melanizzazione o le dimensioni dei melanosomi.
Il termine Albinismo Oculocutaneo (OCA) distinguele condizioni caratterizzate da ridotta sintesi di melanina nella cute, nei capelli e negli occhi (il grado di melanizzazione è variabile: da melanociti amelanotici a melanociti quasi normalmente pigmentati), mentre il termine Albinismo Oculare (OA) distingue quelle caratterizzate da una ridotta sintesi di melanina fondamentalmente nell’epitelio pigmentato della retina oculare, anche se l’analisi istologica, rilevando la presenza sistemica di macromelanosomi, indica il coinvolgimento di tutti i tessuti interessati.
I cambiamenti oculari, di entità variabile e non tutti sempre presenti, si ritrovano in entrambe le forme di Albinismo
E’ da notare che la presenza di ipopigmentazione cutanea o oculare, il colore della cute o dei capelli, o la presenza o assenza della capacità di abbronzarsi non sono sufficienti per definire il tipo di albinismo. Per esempio, individui con alcuni tipi di OCA possono presentare una pigmentazione cutanea relativamente normale, con la possibilità di abbronzarsi fin dall’adolescenza, suggerendo una diagnosi di OA; eppure l’analisi molecolare mostra chiaramente che essi presentano un tipo di OCA pigmnetato.
Fenotipo Albinotico
IL Fenotipo Albinotico presenta una eterogeneità clinica molto estesa, con manifestazioni oculari e/o cutanee che non sono, in generale, né essenziali , né specifiche dell’Albinismo. Sembra possa considerarsi caratteristica specifica dell’Albinismo, indipendentemente dal genotipo e dal fenotipo, il ritrovamento di un unico modello di organizzazione anomala della via visiva (°Apkarian, 1992), elemento molto utile, accanto all’ipoplasia foveale, per un indagine diagnostica, anche in famiglie che presentano espressione variabile di Albinismo tra i propri membri (*Summers, 1996; Castronuovo, 1991; Summers, 1991).
Fenotipo pigmentario albinotico.
● La cute si presenta normale da un punto di vista anatomico-funzionale. L’epidermide ha i suoi tre tipi fondamentali di cellule (cheratinociti, melanociti, cellule di Langerhans) ed il ricambio cellulare è fisiologico. Cambia solo il colore, che copre uno spettro pigmentario estremamente ampio: da un fenotipo apigmentato ad un fenotipo quasi normalmente pigmentato, tanto da risultare, talvolta, non diagnosticabile. In relazione alla quantità di pigmento presente, varia la risposta della cute al sole, ossia l’entità dello schermo di protezione naturale e, di conseguenza, il rischio di pachidermia, cheratosi solare e cancro cutaneo.
● I capelli hanno una normale struttura anatomica, varia solo il colore: dal bianco al giallo chiaro, alle diverse tonalità di biondo e di marrone.
● Il colore dell’iride varia dal blu/grigio opaco al verde/verde nocciola al marrone/marrone chiaro, in relazione alla quantità di pigmento presente nello stroma irideo.
Fenotipo oftalmologico albinotico.
Le anomalie oculari albinotiche (tabella 1), di cui bisogna tener conto per la definizione e per la diagnosi di Albinismo, non sono clinicamente osservabili negli eterozigoti per OCA autosomico recessivo, che sono asintomatici, mentre l’80-90% delle femmine portatrici, eterozigoti obbligate per OA recessivo X-linked, presenta cambiamenti osservabili nella pigmentazione oculare.
Tabella 1. Anomalie oculari nell’Albinismo.
| ● ipopigmentazione dell’iride (stroma; IPE) | trasparenza dell’iride | fotofobia |
| microscopio slit-lamp | ||
| condizioni di luce | ||
| ● ipopigmentazione della retina (RPE) | trasparenza della retina | vasi corioidei visibili |
| riflessione della luce | ||
| ERG normale | ||
| ● ipoplasia della fovea | r./a. riflesso foveale | r. acuità visiva |
| OCT indicativo | ||
| ● percorso anomalo dei nervi ottici al chiasma | r. fibre RGC ipsolaterali | r./a. stereoacuità |
| VEP indicativo | ||
| MRI indicativo | ||
| ● nistagmo | orizzontale | Riduzione acuità visiva |
| pendolare/a scatti | ||
| movimento/inclinazione del capo | ||
| ● strabismo alternante | intermittente | no ambliopia |
| verticale/orizzontale | ||
| ● errori di rifrazione | miopia | |
| iperopia | ||
| astigmatismo |
Nota. r., riduzione; a., assenza; RGC, retinogeniculocorticali; slit-lamp., lampada a fessura.
Da R. Pellegrino, 2007
● Ipopigmentazione dell’iride (fig. 1). Un’ iride blu, ma opaca, non è albinotica. Un’ iride blu e trasparente può essere albinotica. Quando ad un’ ipopigmentazione stromale si aggiunge un’ipopigmentazione dell’epitelio pigmentato dell’iride, il risultato è una trasparenza di grado variabile: da uno stato di completa trasparenza, per cui l’iride appare “rossa” ad occhio nudo o sotto certe condizioni di luce (è il riflesso rossastro o blu violetto che, proveniente dalla retina, attraversa un’ iride molto poco pigmentata e, talvolta, anche la pupilla), ad aree punteggiate di trasparenza (macchie o strisce peripupillari). Il microscopio con la lampada a fessura è lo strumento più comunemente usato per accertare la trasparenza iridea, anche nei casi in cui essa non risulti evidente: i raggi incidenti di un sottile fascio di luce, che attraversa la pupilla non dilatata, riflessi dalla retina, passano non solo attraverso la pupilla, ma anche attraverso l’ride, evidenziandone i difetti di transilluminazione. Un iride con aree di trasparenza non è necessariamente albinotica.
L’iride delle femmine portatrici di OA1, presenta, nell’80-90% dei casi, aree di trasparenza.
Un iride trasparente permette alla luce dispersa di entrare e alla luce riflessa dalla retina di uscire, causando ipersensibilità patologica alla luce: fotofobia (o fotoavversione o fotodisforia), sintomo di intensità variabile, talvolta estremamente invalidante.
La funzione diaframmatica dell’iride non è compromessa.
Fig.1. Iride albinotica (momentaneamente non disponibile)
Osservazioni al microscopio con lampada a fessura.
A. completa transilluminazione dell’iride dovuta all’assenza della melanina.
B. minima transilluminazione dell’iride, dovuta alla presenza di una marcata quantità di pigmento melanico nell’epitelio pigmentato dell’iride.Da Summers, 1996.
● Ipopigmentazione della retina (fig. 2).La retina albinotica, ad un esame oftalmologico, si presenta rosa/rossastra: la sua trasparenza, determinata dalla riduzione/assenza di melanina nell’epitelio pigmentato della retina, rende visibili i vasi sanguigni corioidei sottostanti. L’elettroretinogramma (ERG) è normale: indica che la retina è in grado di ricevere e processare la luce e non documenta deterioramento retinico correlato agli effetti tossici dell’esposizione alla luce, sebbene il filtraggio della luce risulti ridotto.
La retina delle femmine portatrici di OA1 X-linked (eterozigoti obbligate) mostra, nell’80-90% dei casi, i “segni del portatore” (fig.3), un modello, facilmente riconoscibile, di depigmentazione irregolare del fondo oculare: una tessitura granulare nella regione maculare che assume, gradualmente, verso la periferia,un aspetto a strisce depigmentate. Tale mosaico di depigmentazione retinica è stato descritto per la prima volta nel 1951 da Falls (Falls, 1951) e attribuito dallo stesso, e più tardi (1961) da Mary Lyon, alla espressione random delle due X in una femmina.
Fig. 2. Retina albinotica (momentaneamente non disponibile)
Fig. 3. Retina di una femmina portatrice di OA1 X-linked (momentaneamente non disponibile).
Fotografia del fondo dell’occhio di una femmina portatrice. Spiegazione nel testo.Da “The Vision of Children”
● Ipoplasia della fovea (fig.2). La fovea albinotica è ipoplastica, con un riflesso foveale ridotto o assente. L’ispezione del fondo oculare con l’oftalmoscopio mostra l’assenza del riflesso annulare e foveale nella macula, e, talvolta, la presenza di in intreccio di vasi retinici in quest’area normalmente avascolarizzata. L’esame istopatologico degli occhi di soggetti XLOA e OCA1A (*Summers, 1996; Fulton, 1978; O’Donnell, 1996 ) rivela la mancata differenziazione della fovea, l’assenza di una zona maculare priva di bastoncelli, una riduzione nel numero dei coni della retina centrale e l’assenza dei tipici coni cilindrici foveali (fig. 3.4). Taylor (Taylor, 1978) parla di un ispessimento dello strato delle cellule gangliari nell’area foveale, la cui presenza <<provocherebbe un’alterazione dell’immagine retinica>> e di conseguenza un <<difetto non correggibile nella fissazione centrale>>. Un esteso ispessimento della retina, coprente l’intera area foveale, altrimenti depressa, tanto da risultare indistinguibile dalla macula circostante, viene individuato dall’OCT (Tomografia della coerenza ottica) in una bimba di 10 anni con OCA (°Mayer, 2002): lo spessore della fovea è di 300 µm nella paziente contro i 150 µm in un soggetto normale. L’OCT mostra, nella localizzazione anatomica della fovea, un segnale altamente riflettente della retina interna, probabilmente coerente con la presenza di molteplici strati di cellule gangliari, dove non dovrebbero essercene affatto, a conferma dell’ipoplasia della fovea nella paziente esaminata. Tale architettura, viene rivelata anche dai dati OCT di un paziente, esaminato da Recchia e collaboratori (Recchia, 2002) che preferiscono usare il termine di disgenesia foveale, per indicare una fovea più spessa del normale. Sembra che l’OCT consenta un esame dettagliato dell’anatomia della macula nei pazienti con ipoplasia foveale, documentando, in vivo, la morfologia e lo spessore della fovea.
Lo sviluppo anomalo della fovea appare collegato alla riduzione/assenza della melanina durante lo sviluppo dell’occhio (fasi prenatale e postnatale). Sembra che la formazione dell’epitelio pigmentato della retina cominci nelle vicinanze (?) della fovea e proceda, di qui, verso la periferia, con modalità e velocità diverse , suggerendo che la presenza o l’assenza della melanina in queste cellule potrebbe interferire con lo sviluppo della fovea in modo diverso che con la restante parte della retina (°Ilia, 1996).
La fovea ipoplastica causa la riduzione dell’acuità visiva, non correggibile con l’uso di lenti. E’ interessante notare che, nonostante l’invariabile (costante) presenza dell’ipoplasia foveale, l’acuità visiva varia da 20/20 a 20/400. Non intacca invece la visione dei colori, che, nell’albinismo, risulta normale.

Da sinistra a destra: bastoncelli; coni; coni cilindrici della fovea.
Da Bowling J., 1987
● Percorso anomalo dei nervi ottici al chiasma (fig. 5).
Sembra sia l’unica anomalia oculare specifica dell’Albinismo, senza la cui evidenza clinica (stereoacuità ridotta) o elettrofisiologia (VEP asimmetrico) non è possibile fare diagnosi di Albinismo, soprattutto nei casi dubbi. Le fibre nervose della retina temporale (il 20° posteriore o più della retina temporale), giunte a livello del chiasma ottico, decussano e si proiettano al nucleo genicolato controlaterale, piuttosto che al nucleo genicolato ipsolaterale, determinando una riduzione delle fibre che si proiettano ipsolateralmente, una disorganizzazione e frammentazione del modello laminare dei nuclei genicolati dorso-laterali (LGNs) ed una disorganizzazione delle proiezioni dirette da questi nuclei alla corteccia visiva. Ne segue che l’imput visivo dell’occhio destro è quasi esclusivamente diretto verso l’emisfero sinistro e viceversa, per cui non si ha la sovrapposizione dei campi visivi (l’emisfero sinistro è interessato solo al campo visivo di destra), con conseguente riduzione della funzione binoculare (poche fibre retinogeniculostriate si portano comunque ipsolateralmente), necessaria per la visone spaziale (stereoscopica), che risulta quindi ridotta o assente. La tecnica, finora più idonea al rilevamento dell’ anomala distribuzione topografica delle proiezioni retinogeniculocorticali, è la VEP(potenziale evocato visivo), consistente nel posizionare, sull’area occipitale del soggetto in esame, elettrodi registranti, che consentono di individuare le asimmetrie interemisferiche, in seguito alla stimolazione monoculare. L’ampiezza della risposta risulta sproporzionatamente più grande nell’emisfero controlaterale all’occhio stimolato, piuttosto che quasi uguale per ogni emisfero. Gli stimoli pattern (immagini con una “trama” e un “disegno” regolari, ad esempio una scacchiera luminosa) si sono rivelati i più efficaci nell’individuare il “misrouting” nell’Albinismo, quando messi a confronto con altre condizioni di stimolo (°Creel, 1981). Tenuto conto, inoltre, che il profilo VEP è età-specifico (può essere, infatti, utilizzato anche per seguire lo sviluppo e la funzionalità delle vie visive dopo la nascita), il test è stato affinato nel corso del tempo, mettendo in atto procedure appropriate all’età (stimoli flash per i neonati, stimoli pattern onset per i più grandi) ed alla quantificazione dell’estensione dell’anomalia (°Apkarian, 1992;° Hoffmann, 2005), così da fornire una individuazione, quanto più attendibile possibile, del misrouting VEP albino, soprattutto in casi dubbi, ed una diagnosi differenziale definitiva. Alcuni articoli riportano, ad esempio, che il Nistagmo congenito (CN) mostra un’anomalia albino-simile della via visiva. Esaminata la topografia VEP in 10 pazienti con CN, in 10 pazienti con Albinismo e nistagmo ed in 8 pazienti con Albinismo senza nistagmo, si è giunti ad un risultato chiaro: l’asimmetria VEP controlaterale che riflette il misrouting retinico temporale si evince solo nei pazienti albini. Inoltre il CN sembra non possa essere attribuito al misrouting. (°Apkarian, 1991). In uno studio successivo (°Pott, 2003), il valore del coefficiente chiasmatico di Pearson (CC), che si calcola correlando le differenze di potenziale interemisferiche per ogni occhio, ha confermato i risultati precedenti: un CC positivo in 20 individui del gruppo di controllo e in 4 con CN indica una lateralizzazione sullo stesso emisfero, mentre un CC negativo in 6 individui albini indica una lateralizzazione sull’emisfero opposto.
Studi di immagini di risonanza magnetica (MRI) rivelano che le dimensioni e l’ architettura (configurazione) del chiasma ottico nei soggetti albini sono diverse da quelle nei soggetti normali di controllo (fig. 6, fig. 7), rispecchiando l’incrocio atipico delle fibre ottiche nell’albinismo (*Schmitz, 2003). Questo approccio non è stato però ancora sufficientemente testato da poter sostituire la VEP.
L’origine del misrouting sembra correlato alla riduzione/assenza della melanina nell’occhio, durante le prime fasi dell’embriogenesi (primo trimestre di gravidanza). La melanina (topografia e tempo di comparsa) potrebbe avere un ruolo diretto nello sviluppo della proiezioni ottiche, influenzandone la direzione (°Silver, 1981), se non addirittura le proporzioni di cellule retiniche gangliari che si incrociano al chiasma (°Marcus, 1996). Studi recenti suggeriscono che i geni responsabili dell’Albinismo alterino lo sviluppo dei nervi ottici, agendo sullo sviluppo della retina, piuttosto che sulle fasi dello sviluppo dei nervi ottici (°Mason, 1998). L’introduzione del gene della tirosinasi in topi e conigli generati da ceppi albini corregge il misrouting ottico, normalmente osservato in questi animali (Jeffery,° 1997,* 2004).
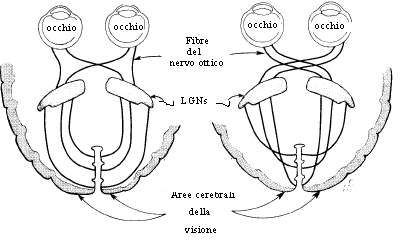
Fig. 5. Percorso delle fibre del nervo ottico.
In un soggetto normale (a sinistra) e in un soggetto albino (a destra).Spiegazione nel testo.Da King, Facts about Albinism (modificata. Pellegrino R, 2007).
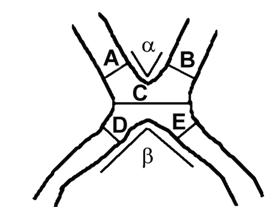
I diametri dei nervi ottici sono stati misurati perpendicolarmente all’asse lungo del nervo, con il punto di inizio mediale 2 mm anteriore al bordo frontale del chiasma ottico (linee A e B).L’ampiezza del chiasma ottico è stata misurata a livello del suo lato più piccolo (linea C).I diametri dei tratti ottici sono stati misurati a 2 mm posteriori al bordo occipitale del chiasma ottico, perpendicolarmente al tratto ( linee D ed E ).Gli angoli tra i nervi ottici sono stati misurati disegnando linee lungo il centro dei nervi ottici e prendendo l’angolo al punto di incrocio di queste due linee. Gli angoli tra i tratti ottici sono stati ottenuti allo stesso modo.Gli angoli sono stati contrassegnati con alfa e beta.
| Gruppo albino (17) | gruppo di controllo (15) | |
| Diametro medio del nervo ottico destro | 4.5 mm | 5.2 mm |
| Diametro medio del nervo ottico sinistro | 4.6 mm | 4.9 mm |
| Ampiezza del chiasma ottico | 10.3 mm | 12.9 mm |
| Diametro del tratto ottico destro | 3.3 mm | 3.7 mm |
| Diametro del tratto ottico sinistro | 3.3 mm | 3.7 mm |
| Angolo tra i nervi ottici (alfa) | 78 gradi | 69 gradi |
| Angolo tra i tratti ottici (beta) | 94 gradi | 79 gradi |
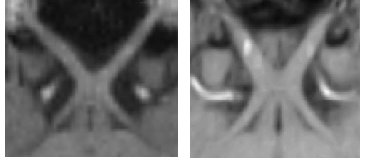
(Le immagini di risonanza magnetica sono state trasferite ad un computer workstation e sono state ottenute immagini riformattate parallele ai nervi e ai tratti ottici e attraverso il chiasma ottico.). A. .Immagine riformattata lungo i nervi e i tratti ottici attraverso il chiasma ottico in un soggetto con albinismo, che mostra un chiasma ristretto e angoli più grandi tra i nervi e i tratti ottici.B. Immagine riformattata lungo i nervi e i tratti ottici attraverso il chiasma ottico in un soggetto normalmente pigmentato, che mostra un chiasma ampio e angoli piccoli tra i nervi e i tratti ottici.
Da Schmitz, 2003.
● Nistagmo congenito.
Osservabile ad un esame ad occhio nudo, è un movimento orizzontale involontario degli occhi che compromette la qualità della visione. Di tipo pendolare o a scatti, è a volte associato ad un movimento compensatore del capo ( ciondolare del capo) o ad una posizione compensatrice del capo (inclinazione del capo), in grado di ridurre l’entità del movimento oculare, grazie al raggiungimento del punto nullo (posizione dello sguardo in cui il nistagmo è meno severo), e quindi il movimento continuo dell’immagine retinica, migliorando, di conseguenza, l’acuità visiva. Presente in quasi tutti gli albini, si manifesta, di solito, nei primi tre mesi di vita e può essere preceduto da un periodo di scarsa fissazione e scarso contatto visivo. In pochi casi è presente alla nascita. Inizialmente molto veloce, tende a diminuire con l’età, ma di rado scompare completamente. E’ più marcato quando il soggetto è triste, arrabbiato o ansioso, meno evidente quando il soggetto è tranquillo. Il soggetto che lo manifesta non ne avverte comunque la presenza, né nota alcun costante movimento nella visione. Il meccanismo responsabile del nistagmo non è ancora chiaro. Il fatto che esso si presenti poco dopo la nascita, momento in cui la fovea normalmente pigmentata sta maturando, suggerisce che la mancanza della normale funzione foveale non abbia un ruolo critico nello sviluppo del nistagmo (°Collewijn, 1985). E’ possibile che questo ruolo sia assunto invece dall’anomalia del percorso dei nervi ottici (Apkarian ne dimostra il contrario; °Apkarian, 1991) o che tutti i cambiamenti oculari albinotici possano contribuire al suo sviluppo (* King, 2001), come del resto contribuiscono alla riduzione dell’acuità visiva (in questo caso però l’ipoplasia foveale è il principale fattore limitante).
● Strabismo alternante. Suggerisce, insieme alla riduzione della visione stereoscopica, la presenza del “misrouting” delle fibre ottiche. Consiste nella soppressione, intermittente ed alternante, della visione in ogni occhio. Con deviazioni sia verticali che orizzontali, non sviluppa ambliopia e non richiede generalmente (se non per motivi estetici), correzione chirurgica.
● Errori di rifrazione. Frequente è la presenza di miopia o iperopia e astigmatismo (nel range di 10 diottrie o più). Lo sviluppo corretto della rifrazione dell’occhio e la crescita postnatale dell’occhio sembra siano regolate dalla qualità delle immagini ricevute dalla retina. Quando la retina riceve immagini defocalizzate l’occhio tenta di compensare cambiando la forma del globo oculare (allungando o accorciando la lunghezza focale) per migliorare la qualità dell’immagine. Il tentativo generalmente fallisce, ma l’occhio alla fine della crescita conserva la forma anomala precedentemente assunta, che determina significativi errori di rifrazione.
Genotipo albinotico.
L’eterogeneità fenotipica è associata ad eterogeneità genica e ad eterogeneità allelica.
La tabella 2 mostra quali sono i geni della pigmentazione, finora associati all’Albinismo.
L’individuazione di soggetti clinicamente albini in cui non è stata rilevata alcuna mutazione in nessuno dei geni noti essere associati all’albinismo, porta a supporre la presenza di mutazioni in regioni regolative o introniche, non ancora esplorate, degli stessi, o l’esistenza di altri geni, non ancora noti, associati a tale fenotipo.
Tabella 2.Geni della pigmentazione associati all’Albinismo.
|
||||||||||||||
|
BIBILOGRAFIA. King. R. A. Facts about of Albinism. Clinical spectrum of Albinism.
Apkarian P et al. VEP projections in congenital nystagmus; VEP asymmetry in albinism: a comparison study. Invest Ophthalmol Vis Sci. 32(9): 2653-2661. 1991. .
Apkarian P. A practical approach to albino diagnosis. VEP misrouting across the age span. Ophthalmic Paediatr Genet. 13(2): 77-88. 1992.
Collewijn H., Apkarian P. et al. The oculomotor behaviour of human albinos. Brain, 108: 1-28, 1985.
Creel D. et al. Evoked potentials in albinos: efficacy of pattern stimuli in detecting misrouted optic fibers. Electroencephalogr. Clin. Neurophysiol. 52(6): 595-603. 1981. .
Falls H. F. Sex-linked ocular albinism displaying typical fundus changes in the female heterozygote. Am. J. Ophthalmol. 34: 41-50. 1951.
E. Giménez et al. A transgenic mouse model with inducibile tyrosinase gene expression using the tetracicline (tet-on) system allows regulated rescue of abnormal chiasmatic projections found in albinism. 2004 Full Text (pdf di 462KB)
Hoffmann MB. et al. Misrouting of the optic nerves in albinism: estimation of the extent with visual evoked potentials. Invest Ophthalmol Vis Sci. 46(10): 3892-3898. 2005.
Jeffery G. et al. Correction of retinal abnormalities found in albinism by introduction of a functional Tyrosinase gene in transgenic mice and rabbits. Devel Brain Res 99: 95-102. 1997.
Marcus RC et al. Retinal axon divergence in the optic chiasm: midline cells are unaffected by the albino mutation. Development 122: 859-868. 1996.
Mason CA and Sretavan DW. Glia, neurons and axon pathfinding during optic chiasm development .Crr Opin Neurobiol. 7: 647-653. 1998.
Meyer C.H. et al. Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence tomography. Am. J. Ophthal. 133: 409-410. 2002. .
Pott JW. et al. Chiasmal coefficient of flash and pattern visual evoked potentials for detection of chiasmal misrouting in albinism. Doc. Ophthalmol. 106(2): 137-143. 2003. Recchia F.M. et al. Optical coherence tomography in the diagnosis of foveal hypoplasia. Arch. Ophthal. 120: 1587-1588. 2002.
B. Schmitz et al. Configuration of the optic chiasm in humans with albinism as revealed by magnetic resonance imaging. 2003. Full Text (pdf di240KB)
Summers, C. G. Vision in Albinism. Trans Am Ophthalmol Soc.94: 1095-1155.1996. Full Text (pdf di 6804KB)
Silver J., Sapiro J. Axonal guidance during development of the optic nerve: the role of pigmented epithelia and other extrinsic factors. J Comp Neurol 202: 521-538. 1981.
Taylor W. O.G. Visual disabilities of oculocutaneous albinism and their alleviation. Trans. Ophthal. Soc. U. K. 98: 423-445. 1978.
NOTA dell'autore.
Mi sia concessa una nota "singolare":
Questo contenuto è stato pubblicato per la prima volta il 4 Ottobre 2007 (troverete la stesura relativa a tale data nella sezione ARCHIVI, con il titolo "Albiniso. Fenotipo e Genotipo - 4 Ottobre 2007: il copia-incolla non aiuta la scienza!"
Questo contenuto è stato oggetto di un letterale copia-incolla
La data della pubblicazione è molto importante: dà ai lettori la possibilità di esprimere un parere sulla correttezza scientifica di un evento scientifico pubblico tenutosi nel 2009 - i cui atti sono stati pubblicati in rete -.
Ai lettori la libertà di mettere o meno in discussione la validità e serietà scientifica di tale evento.
Il termine "scientifico" è volutamnete ridondante: è giusto pretendere correttezza scientifica e professionale anche in rete!
Riportiamo alcuni esempi di copia-incolla
Gradirei una vostra opinione.
Grazie
Rosa Pellegrino
